

迄今最大規模乳腺癌蛋白質組學分析:發現新亞型 提出新療法
發布時間:
2020-11-21 09:00
來源:

近日,貝勒醫學院、麻省理工學院和哈佛大學等機構的研究人員通過強大的蛋白質組學分析,深入了解了乳腺癌的生物學復雜性。在此基礎上,他們能夠對已知的靶標提出更精確的診斷方法,開發侵襲性腫瘤的新治療方法,并揭示乳腺癌耐藥性的新機制。
??該研究于11月18日發表在《Cell》雜志,題為“Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy”。

??蛋白質組學分析將下一代DNA和RNA測序技術與質譜分析相結合,并用計算方法對數據進行綜合處理,從而對癌細胞中的蛋白質和蛋白質修飾進行深入、無偏移的定量分析。先前,美國國家癌癥研究所臨床蛋白質組學腫瘤分析協會(NCI-CPTAC)的研究人員已將這種蛋白質組學方法廣泛應用于癌癥研究。
??在該研究中,研究人員表示:“重要的是,我們的分析包括磷酸化和乙酰化的鑒定,以揭示單個蛋白質活性信息的蛋白質修飾。先前在乳腺癌中未進行蛋白質乙酰化的研究。這些新方法有望提供新的生物學見解,以解決難于治療的乳腺癌及其反應的異質性。”
??同時分析遺傳密碼的變化以及由此產生的蛋白質功能改變,比孤立地分析每個成分,更全面地了解乳腺癌腫瘤內部的狀況。
??更精確的數據
??研究人員使用“癌癥基因組圖譜”中的殘留樣品對乳腺癌進行了初步的蛋白質組學分析,為蛋白質組學代表乳腺癌圖譜研究的進展提供了理論依據。目前的研究向前邁出了的重要一步,它包括使用特定于保留蛋白質修飾的協議收集的組織樣本,分析更多的樣本,對完全相同的組織片段進行基因組學和蛋白質組學鑒定,并在蛋白質磷酸化、DNA和RNA測定中加入蛋白質乙酰化圖譜。近年來,蛋白質基因組分析技術已取得了長足的發展,這些前沿的方法也被應用到了該數據集中。
??研究人員完成了122個治療初期乳腺癌樣本的蛋白質基因組分析。他們的分析產生了大量的數據,包括每個腫瘤內約38000個蛋白質磷酸化位點和近10000個乙酰化位點,以及全外顯子和RNA測序,這些都需要先進的計算方法來分析和整合這些信息。研究人員表示:“如今,這類復雜的分析現在通常在大規模蛋白質組學數據集上進行,并且我們正在開發工具使這一過程自動化。”
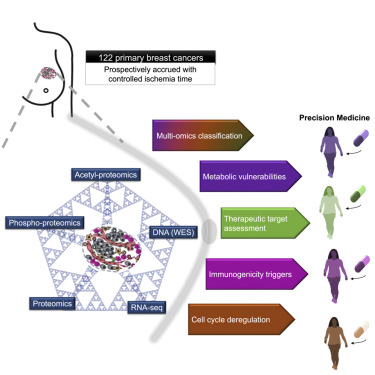
??研究人員說:“在這里,我們描述了迄今為止最大的一組乳腺癌樣本的蛋白質基因組特征,這些樣本是專門為這些類型的分析而收集的,最大限度地提高了結果的保真度和準確性。每個腫瘤細胞實際上都有數百個基因組變化。大多數情況下,我們不了解它們在臨床或生物學上的意義。我們所闡述的方法能夠更深入、更全面地了解每個患者的乳腺癌。”
??確定藥物靶標
??例如,分析顯示,一些乳腺癌亞型有一些被稱為激酶的靶向酶,比其他癌癥的磷酸化程度更高,這表明它具有更高的活性,因此更具有靶向性。這些分析包括最近確定的藥物靶點,如CDK4/6及其調控背景,以及作為新免疫治療藥物靶點的程序性細胞死亡受體和配體。綜合分析還發現了一些新的雌激素受體陽性乳腺癌可以用這些藥物治療。這一點很重要,因為目前這些藥物僅限于雌激素受體陰性疾病。
??其他分析對ER +和ER-乳腺癌的代謝脆弱性提出了全新的見解。研究人員表示:“我們對乳腺腫瘤中首個乙酰基蛋白質組的全面分析揭示了乳腺癌亞型特異性代謝的新細節。”
??改善診斷和治療
??研究人員希望他們的發現能激發乳腺癌科學家探索他們在這項研究中發現的新生物學改變的治療或診斷潛力。他們還樂觀地認為,他們的發現將鼓勵人們努力將蛋白質組學轉變為一種可在臨床上常規用于改善診斷和治療的癌癥分析方法。
??研究人員表示:“我們認為,蛋白質組學方法將繼續幫助我們確定新的候選治療靶標,更好地了解乳腺癌和其他癌癥的免疫狀況,深入了解反應和耐藥性,并朝著實現個性化癌癥治療目標的最終進展邁進。科學是強大而令人興奮的,但最終,我們可以提供給患者的才是重要的。”
地址:上海市徐匯區銀都路218號1C座4樓 聯系人: 盛老師
電話:021-68811399 手機:13601846746 郵箱:service@yihaobio.com
手機二維碼
微信二維碼