

癌癥患者死于心血管疾病的風險通常高于一般人群,根據2019年發表在《歐洲心臟雜志》上的一項覆蓋323萬余美國癌癥患者、跨度長達40年的研究結果提示,每10例癌癥患者中,就有1人最終死于心血管病,而因癌癥死亡者其實只有4人。
發布時間:
2020-09-15 09:00
來源:
心腦血管疾病的預防包括一級預防和二級預防,一級預防是指發病前的預防,即無病防病發生;二級預防是為了降低再次發生的危險及減輕致殘率,即患病后防止再發病。最近有項研究通過遺傳學與基因組學的方向研究心血管疾病的治療。
該項研究于9月9日發表于《Cell Reports》,題目為:“ iPSC Modeling of RBM20-Deficient DCM Identifies Upregulation of RBM20 as a Therapeutic Strategy”,研究人員發現:對于家族性擴張型心肌病(DCM)患者,使用ATRA等藥物來增加心臟細胞中RBM20的水平是一種有前途的治療選擇。

圖:文章截圖 來源:Cell Sports
擴張型心肌病(DCM)會引起心力衰竭。DCM的特征是心臟左心室血量(主泵腔)增加,拉伸的心肌也無法泵血,這會導致心律不齊、心臟瓣膜問題,并最終導致心力衰竭。DCM不僅是心力衰竭的主要原因,也是心臟移植的最常見原因。心臟移植僅在心力衰竭晚期經過其他治療選擇和生活方式改變均告失敗時才考慮。
雖然不斷努力改善心臟移植患者術后生存率,但10年生存率還是只有50%。因此,在進行心臟移植之前,如果能有更好的治療選擇,DCM患者的健康將得到顯著改善。
DCM很多是由人的DNA的遺傳變化(突變)引起的。盡管對突變有詳細的了解,但還沒有針對突變的療法。從誘導多能干細胞(iPSC-CMs)制造心肌細胞的能力進行建模,使人們對遺傳性心臟病有更好理解,并為之尋求治療方法。
由Francesca Briganti博士、Han Sun博士以及資深作家Mark Mercola博士、Lars Steinmetz博士和斯坦福大學的Ioannis Karakikes博士領導的一組研究人員,結合使用iPSC-CM和基因組編輯來識別導致DCM的新突變,從而發現DCM更好的治療。
研究人員對繼承了DCM的家庭進行研究,以了解其病因。作為遺傳病,基因組信息或完整的DNA和基因信息可以提供有關導致疾病突變的重要信息。每個基因都包含制備細胞所需的特定蛋白質,并且該基因從親本傳遞給孩子,包括蛋白質變化,導致疾病如遺傳的DCM。
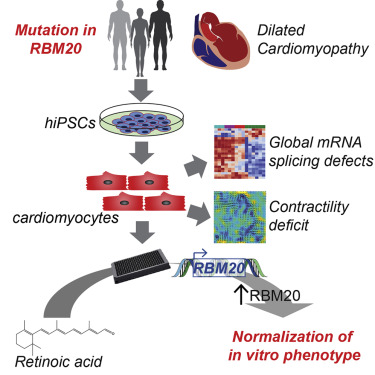
圖 家庭遺傳性擴張型心肌病的RBM20有特定變化
在這種情況下,研究小組比較組成蛋白質(外顯子組)的基因組片段。通過研究不同家庭,他們能夠找出因DCM診斷而去世、被診斷為DCM和不受DCM影響的人的外顯子組差異。通過比較,他們可以在蛋白質RBM20中找到單個突變(P633L),它會引起疾病。盡管突變是未知的,但已知RBM20變化會導致DCM的遺傳。研究人員還發現當基因的一個拷貝被改變(突變),而另一個拷貝是正常時,正常的RBM20功能喪失,這被稱為單倍不足。
iPSC尤其是iPSC-CM平臺是研究特定心血管疾病(如遺傳性DCM)的強大工具。研究人員能夠使用基因組編輯(一種對細胞DNA進行特定改變的過程),將RBM20突變(P633L)引入iPSC-CM。這使他們有機會了解突變是如何引起DCM的。
重要的是,iPSC-CM平臺還允許研究人員通過調節RBM20來測試使用全反式維甲酸(ATRA)作為DCM的潛在治療方法。在體內,ATRA由維生素A制成,可幫助細胞生長和發育。有趣的是,ATRA治療可以增加RBM20的水平。更多的RBM20可使功能恢復正常水平,從而治療由突變(單倍劑量不足)引起的遺傳DCM。在這里,研究人員使用具有特定RBM20突變(P633L)的iPSC-CM來證明ATRA可以部分修復改變后的細胞中的缺陷。至關重要的是,研究人員能夠證明,對于家族性DCM患者,使用ATRA等藥物來增加心臟細胞中RBM20的水平是一種有前途的治療選擇。
地址:上海市徐匯區銀都路218號1C座4樓 聯系人: 盛老師
電話:021-68811399 手機:13601846746 郵箱:service@yihaobio.com
手機二維碼
微信二維碼